Données techniques
| Formule | C22H25N7O2 |
||||||
| Poids moléculaire | 419.48 | N° CAS | 952021-60-2 | ||||
| Solubilité (25°C)* | In vitro | DMSO | 83 mg/mL (197.86 mM) | ||||
| Water | Insoluble | ||||||
| Ethanol | Insoluble | ||||||
| In Vivo (Ajoutez les solvants au produit individuellement et dans lordre.) |
|
||||||
|
* <1 mg/ml signifie légèrement soluble ou insoluble. * Veuillez noter que Selleck teste la solubilité de tous les composés en interne, et la solubilité réelle peut différer légèrement des valeurs publiées. Ceci est normal et est dû à de légères variations dun lot à lautre. * Expédition à température ambiante (les tests de stabilité montrent que ce produit peut être expédié sans aucune mesure de refroidissement.) |
|||||||
Préparation des solutions mères
Activité biologique
| Description | PF-477736 (PF-736, PF-00477736) est un inhibiteur sélectif, puissant et ATP-compétitif de Chk1 avec un Ki de 0,49 nM dans un essai acellulaire et inhibe également VEGFR2, Aurora-A, FGFR3, Flt3, Fms (CSF1R), Ret et Yes. Il montre une sélectivité ~100 fois supérieure pour Chk1 que pour Chk2. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cibles |
|
||||||||||
| In vitro | PF-477736 (128 nM) abroge le point de contrôle des dommages à l'ADN induits par la camptothécine de manière dose-dépendante dans les cellules CA46 et HeLa. Ce composé abroge efficacement l'arrêt en phase S induit par avec une augmentation correspondante des populations de cellules apoptotiques dans les cellules HT29. Ce produit chimique (540 nM) améliore la cytotoxicité induite par de manière dépendante du temps et de la dose dans les cellules HT29. Il potentialise l'activité inhibitrice de croissance d'un panel d'agents chimiothérapeutiques sur un large spectre de lignées cellulaires cancéreuses humaines déficientes en p53 dans l'essai MTT. L'ajout de ce composé (360 nM) à des cellules arrêtées par induit une augmentation spectaculaire de l'intensité de la phosphorylation de H2AX, reflétant un plus grand nombre de molécules γ-H2AX près des sites de dommages à l'ADN. Ce produit chimique (0,5 nM) bloque sélectivement la phosphorylation de p73 et P53 en présence de curcumine dans les cellules HL-60. Il (360 nM) supprime la phosphorylation induite par de l'histone H3 (Ser10) et de Cdc25C (Ser216) et potentialise l'apoptose dans les cellules COLO205. Ce composé (250 nM) combiné à MK-1775 a une activité cytotoxique synergique marquée dans les cellules OVCAR-5. Il (250 nM) combiné à MK-1775 provoque l'accumulation de cellules avec un contenu en ADN entre 2N et 4N dans les cellules OVCAR-5. Ce produit chimique (250 nM) combiné à MK-1775 provoque une mitose prématurée avant la fin de la réplication de l'ADN, l'ADN endommagé conduisant à la mort cellulaire apoptotique dans les cellules OVCAR-5. |
||||||||||
| In Vivo | Le PF-477736 (4 mg/kg i.v.) entraîne une demi-vie terminale (T1/2) de 2,9 heures, une AUC de 5,72 μg×hr/mL et une CLp de 11,8 mL/min/kg chez les rats. Ce composé améliore de manière dose-dépendante l'activité antitumorale d'une dose maximale tolérée dans le modèle murin de xénogreffe Colo205. Ce produit chimique (12 mg/kg) induit une augmentation de la phosphorylation de l'histone H3 (Ser10) et de la phospho-histone H2AX dans le modèle murin de xénogreffe Colo205. Ce composé (15 mg/kg i.p.) améliore l'inhibition de la croissance tumorale induite et le retard de croissance tumorale dans les modèles de xénogreffe COLO205 et MDA-MB-231. Ce produit chimique (10 mg/kg une fois par jour i.p.) combiné à MK-1775 (30 mg/kg deux fois par jour par voie orale) entraîne une plus grande inhibition de la croissance tumorale chez les souris porteuses de xénogreffes OVCAR-5. |
Protocole (de référence)
| Test kinase : |
|
|---|---|
| Test cellulaire : |
|
| Étude animale : |
|
Références
|
Validation du produit par le client
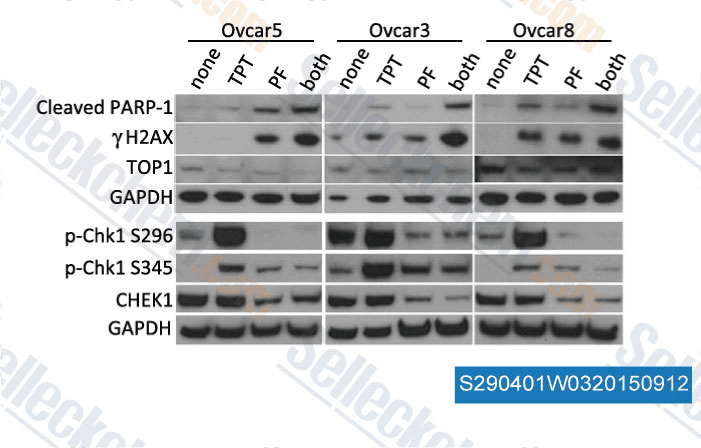
-
Données de [ , , BMC Cancer, 2015, 10.1186/s12885-015-1231-z ]
De Selleck PF-477736 A été cité par 36 Publications
| Synthetic Lethal Combinations of DNA Repair Inhibitors and Genotoxic Agents to Target High-Risk Diffuse Large B Cell Lymphoma [ Hematol Oncol, 2025, 43(5):e70131] | PubMed: 40847617 |
| Centrioles are frequently amplified in early B cell development but dispensable for humoral immunity [ Nat Commun, 2024, 15(1):8890] | PubMed: 39406735 |
| An RNA damage response network mediates the lethality of 5-FU in colorectal cancer [ Cell Rep Med, 2024, 5(10):101778] | PubMed: 39378883 |
| ATR, CHK1 and WEE1 inhibitors cause homologous recombination repair deficiency to induce synthetic lethality with PARP inhibitors [ Br J Cancer, 2024, 10.1038/s41416-024-02745-0] | PubMed: 38965423 |
| A multiparametric screen uncovers FDA-approved small molecules that potentiate the nuclear mechano-dysfunctions in ATR-defective cells [ Sci Rep, 2024, 14(1):30786] | PubMed: 39730498 |
| Mild replication stress causes premature centriole disengagement via a sub-critical Plk1 activity under the control of ATR-Chk1 [ Nat Commun, 2023, 14(1):6088] | PubMed: 37773176 |
| BRD7 suppresses tumor chemosensitivity to CHK1 inhibitors by inhibiting USP1-mediated deubiquitination of CHK1 [ Cell Death Discov, 2023, 9(1):313] | PubMed: 37626049 |
| The p38/MK2 Pathway Functions as Chk1-Backup Downstream of ATM/ATR in G2-Checkpoint Activation in Cells Exposed to Ionizing Radiation [ Cells, 2023, 12(10)1387] | PubMed: 37408221 |
| Overcoming PARP inhibitor resistance by inducing a homologous recombination repair defective phenotype with ATR, CHK1 and WEE1 inhibitors [ bioRxiv, 2023, 10.1101/2023.07.05.547758] | PubMed: None |
| Deregulation and epigenetic modification of BCL2-family genes cause resistance to venetoclax in hematologic malignancies [ Blood, 2022, blood.2021014304] | PubMed: 35704690 |
POLITIQUE DE RETOUR
La politique de retour inconditionnelle de Selleck Chemical garantit une expérience dachat en ligne fluide à nos clients. Si vous nêtes en aucun cas satisfait de votre achat, vous pouvez retourner tout article dans les 7 jours suivant sa réception. En cas de problèmes de qualité du produit, quil sagisse de problèmes liés au protocole ou au produit, vous pouvez retourner tout article dans les 365 jours suivant la date dachat initiale. Veuillez suivre les instructions ci-dessous lors du retour des produits.
EXPÉDITION ET STOCKAGE
Les produits Selleck sont transportés à température ambiante. Si vous recevez le produit à température ambiante, soyez assuré que le service dinspection de la qualité de Selleck a mené des expériences pour vérifier que le placement à température normale pendant un mois naffectera pas lactivité biologique des produits en poudre. Après réception, veuillez stocker le produit conformément aux exigences décrites dans la fiche technique. La plupart des produits Selleck sont stables dans les conditions recommandées.
NON DESTINÉ À UN USAGE HUMAIN, VÉTÉRINAIRE DIAGNOSTIQUE OU THÉRAPEUTIQUE.